Indicaid test
Indicaid Antigen 25 tests box

- SKU:
- Indicaid-test-25
- Availability:
- We ship within 8 hours of receiving your order. Next day delivery of all Panbio Tests except for weekends.
Bulk discount rates
Below are the available bulk discount rates for each individual item when you purchase a certain amount
| Buy 20 - 39 | and get 4% off |
| Buy 40 - 59 | and get 6% off |
| Buy 60 - 79 | and get 7% off |
| Buy 80 - 99 | and get 9% off |
| Buy 100 or above | and get 10% off |














Description
Indicaid Omicron Rapid Antigen Test ( 25 test a box)
- 6.5 USD per test
- Box pricing (25 tests per box): 155 USD
- Case pricing (500 tests | 20 boxes) 3000 USD (discounted to $6.00 per test)
- First used Antigen Testing (Rapid) in USA

Boxes are shippen out from monday till friday for next day delivery
100 boxes means 2500 tests in 5 cases of 20x25tests

note: the test is pronounced as "Indicate Self test" because it helps you to indicate if you are safe or not to other persons or if you have been infected yourself and you should look for support.
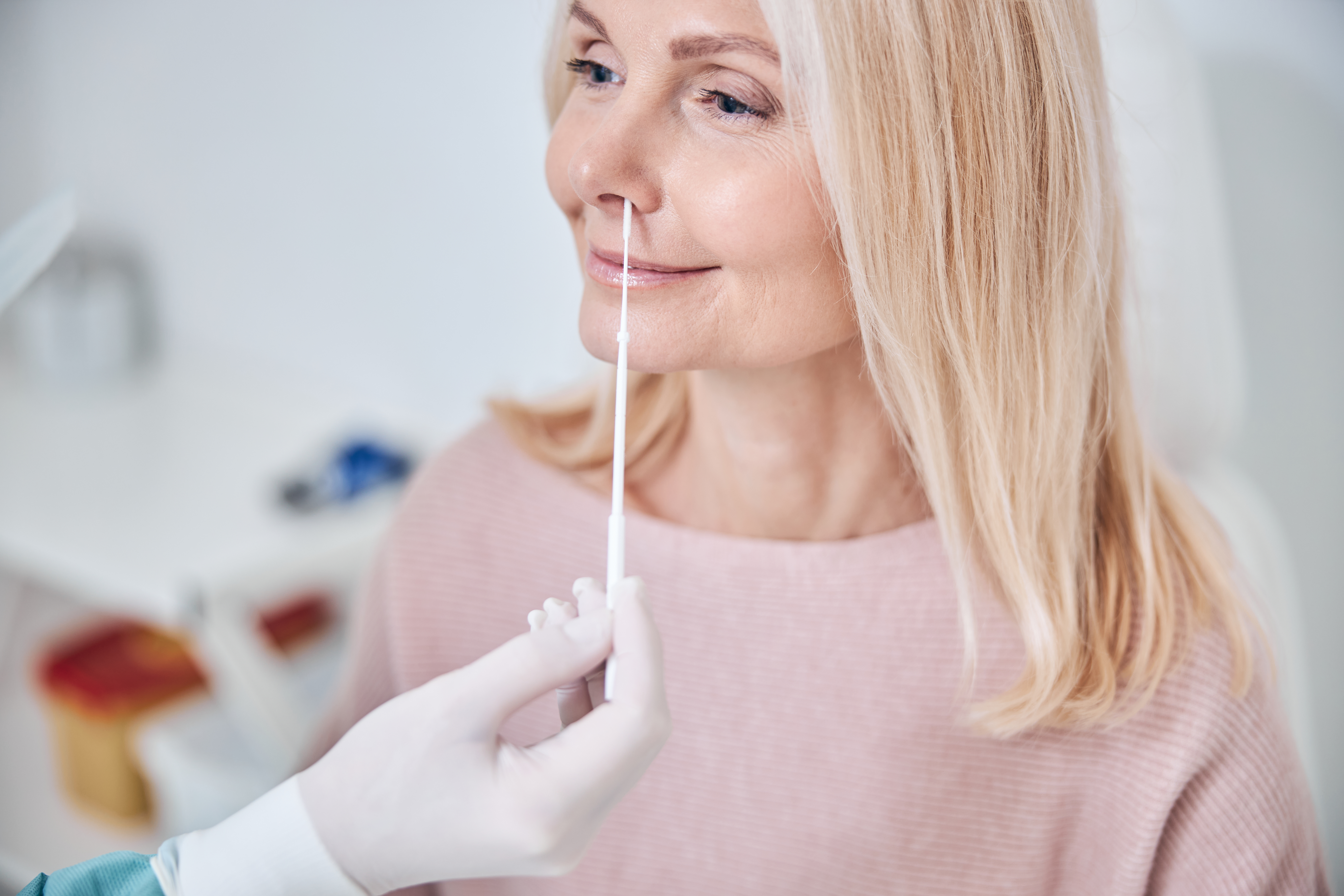
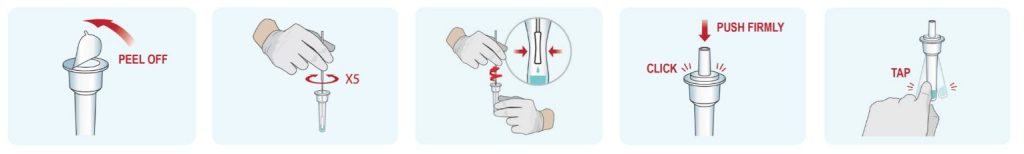
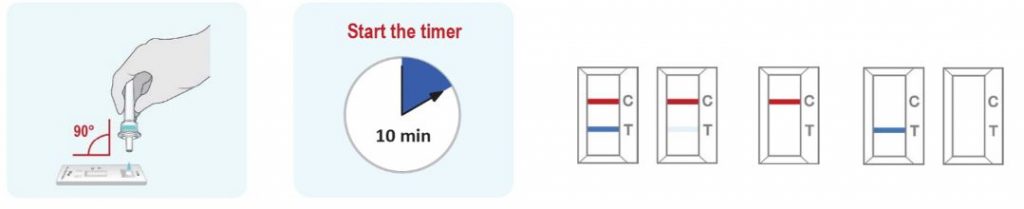
How to use the Indicate test?
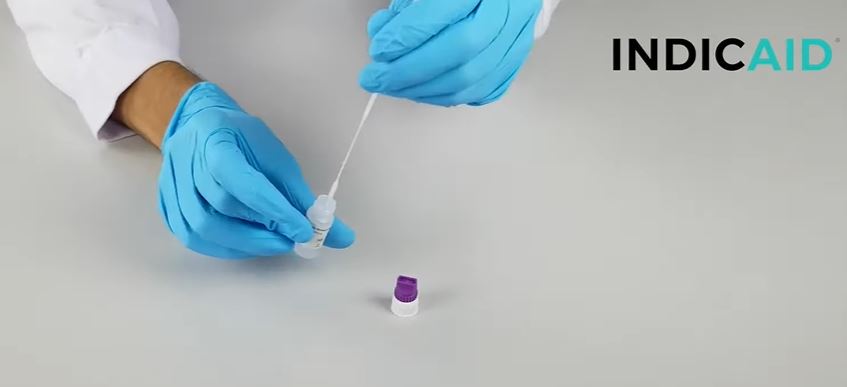
- The kit is very easy to use. The nasopharingeal shwab can be brought less deep into the nose when you exhaust wet air. The liquid should be collected by the shwap.
- Today januray 2022 no saliva test kits are allowed by the FDA but we hope to have soon caliva test kits approved.
- The cassete detects the N capside of the virus that is present on the 2022 strains of Omicron and Delta.
- Results are within 15 minutes and not within 1 hour like for PCR. ( PCR however will detect 2 to 4 days earlier)
FACT SHEET

PHASE Scientific International, Ltd. July 28, 2021
INDICAID™ C-19 Rapid Antigen Test
This Fact Sheet informs you of the benefits of the emergency use of the Atlas Link INDICAID Rapid Antigen Test.
The Atlas Link Rapid Antigen Test is authorized for use using direct anterior nasal swab specimens from within the first five (5) days of signs of acute respiratory illness (e.g., cough, dyspnea) onset.

Current information on COVID-19 for healthcare providers is available at CDC’s webpage, Information for
Healthcare Professionals
• The INDICAID Rapid Antigen Test can be used to test direct anterior nasal swab
specimens. Anterior nasal swab specimens may be
collected by a healthcare provider (HCP) or selfcollected (by individuals 18 years of age or older,
under the supervision of an HCP).
• The INDICAID Rapid Antigen Test should be ordered for the detection in
individuals who are suspected of C-19 by their
healthcare provider within the first five (5) days of
symptom onset.
• The Atlas Link INDICAID Rapid Antigen Test is authorized for use in laboratories certified under the
Clinical Laboratory Improvement Amendments of
1988 (CLIA), 42 U.S.C. §263a, that meet requirements to perform moderate complexity, high
complexity, or waived tests.
• The INDICAID C Rapid Antigen Test is
authorized for use at the Point of Care (POC), i.e., in user care settings operating under a CLIA
Certificate of Waiver, Certificate of Compliance, or Certificate of Accreditation.
• Please refer to the INDICAID Rapid Antigen Test Instructions for Use for additional
information.

- Specimens should be collected with appropriate infection control precautions.
- Current guidance for collecting and handling specimens should be followed for individuals suspected of being infected.
- The CDC Interim Laboratory Biosafety Guidelines for Handling and Processing
Specimens
Rapid Antigen Test
This test is to be performed only using direct anterior nasal swab specimens from individuals by their healthcare
provider.
FACT SHEET FOR HEALTHCARE PROVIDERS
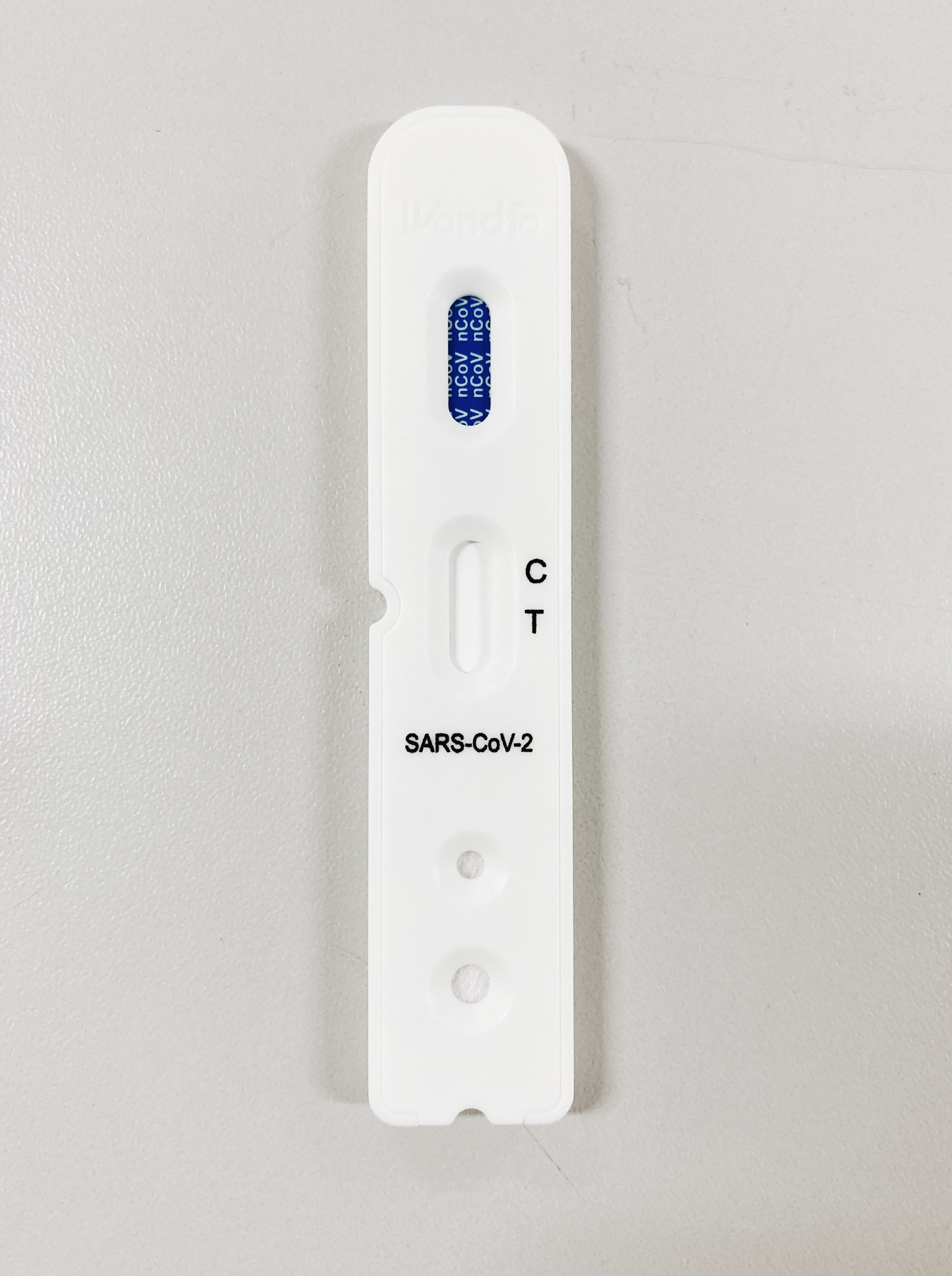
A positive test result indicates that nucleocapsid antigens present in all today known variants including Omicron were detected, and the patient is infected with the virus and presumed to be contagious.
Laboratory test results should always be considered in the context of clinical observations and epidemiological data (such as local Omicron outbreak/epicenter locations)
The INDICAID Omicron Rapid Antigen Test has been designed to minimize the likelihood of false positive
test results.
All laboratories using this test must follow the standard testing and reporting guidelines according to their States
appropriate public health authorities.
What does it mean if the specimen tests negative?
A negative test result for this test means that nucleocapsid antigens from SARS variant were not
present in the specimen above the limit of detection.

However, a negative result does not rule out a latent infection and should not be used as the sole basis for decisions
Antigen tests are known to be less sensitive than molecular PCR tests that detect viral nucleic
acids but they can be done dayly to monitor large groups of persons.
The amount of antigen in a sample may decrease as the duration of illness increases. Specimens collected after
day 5 of illness may be more likely to be negative compared to a RT-PCR assay. Therefore, negative
results from patients with symptom onset beyond 5 days should be treated as presumptive and confirmed with a
molecular assay, if necessary, for patient management.
It is possible to test a person too early or too late to make an accurate diagnosis via the
INDICAID Rapid Antigen Test?
Yes it is absolutely possible when the testing is negative, the possibility of a false negative result should be considered in the context of a patient’s recent exposures and the presence of clinical signs and symptoms.
Additional PCR testing may be helpful to ensure testing was not conducted too early.
Risks to a patient from a false negative result include to early or to late testing.
For additional recommendations regarding infection control, refer to CDC’s Discontinuation of
Isolation

Where do I report Adverse events, including problems with test performance or results?
Your complaints should be reported to to MedWatch by submitting the online FDA Form 3500
(https://www.accessdata.fda.gov/scripts/medwatch/index.cfm?action=reporting.home) or by calling 1-800-FDA-1088
The performance of the Atlas Link Indicate test was established based on the evaluation of a limited number of clinical specimens collected between February 2021 and March 2021. The clinical performance has not been established in all circulating delta and omicron variants but is anticipated to be reflective of the less mutating N capside present in Omicron (B.1.1.529): Variant of Concernfor2022.

What is an EUA?
- The United States FDA has made this test available under an emergency access mechanism called an
Emergency Use Authorization (EUA). - The EUA is supported by the Secretary of Health and Human Service’s (HHS’s) declaration that circumstances exist to
justify the emergency use of in vitro diagnostics (IVDs) for the detection and/or diagnosis of the virus. - An IVD made available under an EUA has not undergone the same type of review as an FDA-approved
or cleared IVD. - FDA may issue an EUA when certain criteria are met, which includes that there are no
adequate, approved, available alternatives, and based on the totality of scientific evidence available, it is
reasonable to believe that this IVD may be effective. - The EUA for this test is in effect for the duration of the declaration justifying emergency use of IVDs,
unless terminated or revoked (after which the test may no longer be used).
What are the approved available alternatives?
There are no approved available alternative antigen tests. Any tests that have received full marketing status
(e.g., cleared, approved), as opposed to an EUA, by FDA can be found by searching the medical device databases here:
https://www.fda.gov/medicaldevices/device-advice-comprehensive-regulatoryassistance/medical-device-databases.
FDA has issued EUAs for other tests that can be found at:
https://www.fda.gov/emergency-preparedness-andresponse/mcm-legal-regulatory-and-policyframework/emergency-use-authorization.

Where can I go for updates and more information?
CDC webpages:
General: https://www.cdc.gov/coronavirus/2019-ncov/index.html
Symptoms:
https://www.cdc.gov/coronavirus/2019-ncov/symptomstesting/symptoms.html
Healthcare Professionals:
https://www.cdc.gov/coronavirus/2019-nCoV/hcp/index.html
Information for Laboratories:
https://www.cdc.gov/coronavirus/2019-nCoV/lab/index.html
Laboratory Biosafety: https://www.cdc.gov/coronavirus/2019-
nCoV/lab-biosafety-guidelines.html
Isolation Precautions in Healthcare Settings:
https://www.cdc.gov/infectioncontrol/guidelines/isolation/index.html
Specimen Collection: https://www.cdc.gov/coronavirus/2019-
nCoV/guidelines-clinical-specimens.html
Infection Control: https://www.cdc.gov/coronavirus/2019-
ncov/php/infection-control.html
FDA webpages:
General: www.fda.gov/novelcoronavirus
EUAs:(includes links to fact sheet for individuals and
manufacturer’s instructions) https://www.fda.gov/medicaldevices/coronavirus-disease-2019-covid-19-emergency-useauthorizations-medical-devices/in-vitro-diagnostics-euas
Limitation of Coronavirus Antigen Rapid Test
- This test is recommended for Healthcare professional use.
- The test procedure, precautions and interpretation of results for this test must be followed strictly when testing. Failure to follow the Test Procedure and Interpretations of Test Results may adversely affect test performance and/or invalidate the Test Result.
- The test should be used for the qualitative detection of SARS-CoV-2 antigen in human nasopharyngeal swab specimens. Neither the quantitative value nor the rate of SARS-CoV-2 antigen concentration can be determined by this qualitative test.
- A negative test result may occur if the level of antigen in a sample is below the detection limit of the test.
- Positive test results do not differentiate between SARS-CoV and SARS-CoV-2.
- Positive test results do not rule out co-infections with other pathogens.
- False-negative test results are more likely during peak activity when prevalence of disease is high.
- False-positive test results are more likely during periods of low SARS-CoV-2 activity when prevalence is moderate to low.
Related Products

Canine Heartworm Antigen | RA-40HRT
Immunology Consultant Laboratory


D-Tec® CB Canine Brucellosis Test Kit - 25 ct
Zoetis

Brucellosis Serum Ab 10 Tests
Gentaur Canine Brucella

miRZip-25 anti-miR-25 microRNA construct
SBI

